Article Text

Abstract
The pathological diagnoses and classification schemes for thyroid carcinoma have changed over the past 20 years and continue to do so. New entities have been described and molecular analyses have suggested better characterisation and grouping of certain tumours. Because some of the lesions have been named differently by different authors, clinicians and patients may be confused as to what a specific patient's lesion represents. In this review, we discuss the thyroid tumours of follicular origin which are clinically unusual but important to recognise as their behaviour may be aggressive, they may not respond to radioiodine treatment and they may cause significant mortality. This paper describes these important but rare lesions, their pathological features, important clinicopathological correlations, molecular correlates and prognostic implications.
- THYROID CANCER
- SURGICAL PATHOLOGY
- NEOPLASMS
Statistics from Altmetric.com
Introduction
The incidence of thyroid carcinomas is increasing globally; in 2012 it is expected to be the fifth leading cancer in women in the USA.1 As per National Cancer Institute data, there will be an estimated 56 460 new cases and 1780 deaths from thyroid cancer in 2012 in the USA.1 ,2 The recent literature on thyroid carcinoma has included many studies on clinical features, pathological subtypes and molecular characteristics. Some pathological studies have added confusion to the literature by classifying certain tumours with different names (eg, insular carcinoma, poorly differentiated carcinoma). In addition, new terminology has been suggested for certain thyroid cancers (‘hobnail’, ‘micropapillary’ carcinoma (the latter being different from papillary microcarcinoma!)). Finally, the pathological classification of follicular patterned tumours of the thyroid has caused confusion among clinicians and patients; the degree of interobserver variability has added to this problem.
In this paper, we review the types of thyroid carcinoma which are associated with aggressive clinical behaviour and list the pathological features of importance to aid in the classification. The implications of these tumours are discussed.
Aggressive variants of papillary thyroid carcinoma
Papillary thyroid carcinoma (PTC) is the most common thyroid malignancy (85% of cases) and usually follows an indolent clinical course with overall 5-year relative survival being 97.5%. Only a small percentage of papillary carcinomas behaves with considerable clinical aggressiveness; these are recognised by WHO as biologically aggressive variants.3 Some experts have referred to these as ‘real carcinomas’ of the thyroid.4 This list of aggressive subtypes of PTC include tall cell, columnar cell, diffuse sclerosing variant and hobnail variant. All have been associated with higher rates of extrathyroidal extension (ETE), multifocality, and nodal and distant metastasis, recurrence, and resistance to radioactive iodine therapy. There has been an increase in incidence of aggressive variants of PTC. A recent study assessing 43 738 patients found an increase in the incidence of diffuse sclerosing variant and tall cell variant by 126% and 158%, respectively compared with a 60.8% increase in classical PTC.2 ,5 ,6
Tall cell variant of PTC
The tall cell variant of PTC (TCV-PTC) is an uncommon tumour, accounting for 1.3–12% of all PTCs.7–9 It is associated with aggressive clinical features and high mortality rates. Compared with classical PTC, TCV-PTC presents at an older age (54.3 years vs 46.3 years), has a higher rate of ETE (53.6% vs 30.2%) and poorer 5-year disease-specific survival (81.9% vs 97.8%). Most patients with TCV-PTC have lymph node metastasis at the time of presentation. These tumours tend to be larger than the classical PTCs; mitotic activity and ETE is easily seen.6 ,8–10
Based on the initial description by Hawk and Hazard TCV-PTC is characterised by a papillary growth pattern and tumour cells which are at least twice as long (tall) as they are wide.11 This description has been modified to tumour cells being three times their width.3 The TCV-PTC usually shows complex papillary architecture comprising of elongated and thin papillae; which when coalesce, simulate a trabecular and/or solid growth pattern on low power microscopic examination. The cells are large, round to oval in shape and often eosinophilic without cytoplasmic granularity; a feature which distinguishes them from true oncocytic follicular/ Hürthle cells.3 ,7 The nuclei are elongated and sometimes conform to the elongated cell in which they are contained, have prominent intranuclear grooves, clearing and intranuclear inclusions7 ,12 (figure 1A). In fine-needle aspiration (FNA) specimens, special attention should be paid to these characteristic cytological features, described by some pathologists as ‘tail-like cells’ or ‘tadpole cells’ Multiple intranuclear inclusions (AKA soap bubble inclusions) may be seen, especially in fine-needle aspiration preparations; these can be helpful in diagnosing this tumour as a tall cell variant of papillary carcinoma13 (figure 1B).

Tall cell variant. Papillary formations are lined by tumour cells with eosinophilic cytoplasm and cell height 2–3 times the width (A— H&E stain, 60×). Fine needle aspiration of tall cell variant of papillary thyroid carcinoma demonstrating ‘soap bubble intranuclear inclusions’ (arrow) (B—Papanicolaou stain, 100×).
Many clinicians feel that there are significant consequences to the pathological diagnosis of TCV-PTC.14 ,15 However, according to the literature there appears to be confusion on the part of pathologists in recognising this tumour or alternatively in overdiagnosing TCV. In our experience the pathology review of these cases often raises the following questions: how much of a tumour needs to show the features of TCV to be diagnosed as such? What is not TCV? What are the consequences of a diagnosis of TCV? And finally, what are the molecular features of this group of tumours and how can they be helpful in understanding the pathogenesis and behaviour of this variant of PTC.
The TCV-PTC should comprise of at least 50% TCV morphology.10 ,12 ,16 The literature is problematic in this regard with TCV diagnosed in tumours having anywhere from 10% to 70% of a particular tumour.16 To further complicate this issue some early publications have included tall cell variant under the heading of poorly differentiated thyroid carcinoma (PDTC).17 It is of particular importance to note that many classic variants of PTC especially the Warthin-like variant will show a minor component of TCV (usually 5–10%).10 These are not to be diagnosed as TCV papillary carcinomas, but this should be mentioned in the pathology report. In our experience, metastases or recurrences of these cases may show a higher percentage of TCV.
In order to isolate true examples of TCV, it is necessary not to include other neoplasms that may superficially resemble this tumour but do not have its prognostic implications. These include Warthin-like papillary carcinoma, oncocytic papillary carcinoma or its follicular variant, oncocytic follicular adenoma (AKA Hürthle cell adenoma) and/or oncocytic adenomatoid nodule with papillary growth pattern.7 There is a general consensus that as a group, TCV has a higher recurrence and death rate than classical PTC. Initially, this was attributed to the fact that TCV presented as large tumours with ETE in older patients.12 However, recent data support that tall-cell histology alone remains a significant prognostic factor independent of age, gender, and tumour size and ETE.14 A meta-analysis of 131 cases of TCV-PTC showed 4.5 times recurrence rate and 14 times greater disease-related mortality as compared with classical PTC.18 It has been suggested that the aggressive behaviour of TCV-PTC is associated with the molecular profile of these tumours.19 The aggressive behaviour of the TCV could be related to certain factors elaborated by the tumour. The high expression of Muc1 and type IV collagenase (matrix metalloproteinase-2) in these TCV-PTCs may allow for degradation of stroma and greater invasive properties.20 ,21 A very significant finding in a majority of (>70%) TCVs tested to date is that they show a point mutation in the BRAF proto-oncogene.22 The aggressive behaviour of TCV-PTC may also be related to the higher prevalence of activating point mutations of the BRAF; as these mutations in PTC have been associated with higher frequency of extraglandular extension and nodal metastases. Loss of heterozygosity for chromosome 1 (D1S243) and the p53 gene (TP53) have been reported though not consistently in TCV-PTC and not in classical PTC, and can be used as a potential tool for molecular discrimination between these two neoplasms.23 In another study, RET/PTC3 rearrangement, which leads to activation of downstream mitogenic signalling pathways, was found in 35.8% of TCVs.24
Finally, the importance of TCV is accentuated by the fact that it is over-represented in those thyroid carcinomas that are refractory to radioactive iodine therapy.12 ,19
In summary, the clinicopathological data available on TCV clearly demonstrates that this is a biologically and clinically aggressive form of PTC. Therefore, it is prudent that presence of any foci of tall cells should be mentioned in a pathology report regardless of the percentage of tall cell cytology found. This should prompt the clinician to fully treat and carefully monitor the patient for recurrence, distant metastasis and transformation to anaplastic carcinoma.
Columnar cell variant
This is a rare subtype with variable biological behaviour, mostly considered as aggressive tumours associated with widespread dissemination and a fatal outcome.25 The most common presenting symptom is an asymptomatic or enlarging neck mass.26 Some consider this variant as a distinct morphological type but not a distinct clinical type of thyroid papillary carcinoma.26 Macroscopically, the tumours can vary from encapsulated to infiltrative. Histologically, the tumours can have diverse growth patterns, including papillary, solid, follicular and trabecular. A common pattern is the presence of markedly elongated follicles arranged in parallel cords (figure 2).26 Evans first described unique histological features of papillary formation, hyperchromatic elongated nuclei and prominent nuclear pseudostratification in two cases of this clinically aggressive thyroid tumour in 1986.27 LiVolsi later described an additional feature of prominent subnuclear vacuolation resembling early secretory endometrium in cases of columnar cell carcinoma.28 This peculiar morphology is also reminiscent of colonic adenomas/adenocarcinomas.29 Currently WHO classification criteria for the columnar cell variant (CCV) of PTC, includes neoplastic cells with elongated nuclei, hyperchromasia, supranuclear and/or subnuclear cytoplasmic vacuolisation, and papillary, follicular, trabecular and/or solid growth patterns.3 CDX2, a nuclear transcription factor important in intestinal development has been shown to be selectively expressed in 50% of cases of CCV.30 ,31 In the past some authors have lumped these tumours with TCV-PTC, however, most experts believe it to be a distinct variant of PTC. It has been proposed that a diagnosis of CCV should only be rendered when a thoroughly sampled tumour contains at least 30% columnar cells.3 It has been shown that CCV is aggressive, due to its rapid growth, high local recurrence rate, and frequent lung, brain and bone metastases. Aggressive surgical and medical management are recommended for these tumours.8

Columnar cell variant. Papillary formation, hyperchromatic elongated nuclei and prominent nuclear pseudostratification (H&E stain, 60×).
The encapsulated variant of CCV also reported by Evans, are thickly encapsulated tumours, which may only show capsular invasion and behave in an indolent fashion.32 Wenig et al26 have reported similar observations. In this study the cases of CCV, which were encapsulated or limited to the thyroid, had favourable clinical course as compared to two cases with extensive extrathyroidal extension. Thus the spectrum of CCV has been broadened; when one includes tumours that are predominantly encapsulated and are confined within the thyroid, the adverse prognosis originally reported does not appear to be present.32 BRAF mutations can be seen in approximately 33% of CCV-PTCs similar to conventional PTC.33
Diffuse sclerosis variant
The diffuse sclerosis variant (DSV) is a rare subtype of PTC which was first described Vickery et al in 1985.34 It represents only about 1.8% of all papillary carcinomas in large series. This tumour, which most often affects children and young adults between 15 years and 30 years of age35 is characterised by diffuse involvement of one or both lobes of the thyroid gland and may present as bilateral goitre.36 ,37 Grossly, the tumour is extremely hard reflecting the extensive calcification.38 ,39 In addition to classic papillary nuclear features, it is histologically characterised by dense sclerosis, extensive squamous metaplasia, focal to diffuse lymphocytic infiltration, numerous psammoma bodies and small papillary to solid tumour deposits within intraglandular lymphatics (figure 3). Due to the presence of numerous psammoma bodies the ultrasound of the thyroid will show lymphatics outlined by calcium AKA ‘snowstorm appearance’.40 The squamous metaplasia of the tumour papillae resembles ‘morular’ metaplasia of the endometrium.4 As stated above, the lymphocytic infiltrates are found around the tumour foci; indeed, the background thyroid shows well-developed chronic lymphocytic thyroiditis (figure 3A,B).41 Fine needle aspirate may show overwhelming presence of slightly atypical monomorphic small lymphocytes that could be misleading. However, the presence of classical papillary nuclear features, with squamous metaplastic epithelium, and abundant psammoma bodies will define this neoplasm42 (figure 3C) The histology and immunoprofile of the background thyroid are identical to Hashimoto's disease. It is unclear if the thyroiditis precedes the tumour or is developed as a reaction to the neoplasm.39 The lesions often show extracapsular extension, distant and nodal metastases (almost 100% will have regional node involvement at presentation) and recur in the neck. Earlier studies have shown that these tumours have a somewhat more serious prognosis than usual childhood/adolescent papillary cancer and a decreased disease-free survival when compared with the usual-type papillary carcinoma.38 ,41 ,43 However, the overall survival is not significantly different. The recent reports and case series have shown that the prognosis of DSV-PTC is as good as that of classic PTC (26) if aggressive treatment is given.44

Diffuse sclerosis variant. Low power showing an infiltrating tumour containing numerous psammoma bodies (arrows) (A— H&E stain, 40×). Fine needle aspiration showing a nest of tumour cells containing a psammoma body (arrow) (B—Papanicolaou stain, 100×).
Abnormalities of E-cadherin/catenin adhesion complex have been identified in the diffuse sclerosing variant and appear to be more pronounced in DSV than in classical PTC.45 In DSV there is a pronounced reduction in its membranous expression, accompanied by a relocation to the cytoplasm compared to classic PTC where there is heterogeneous loss of E-cadherin expression. Inactivation of the E-cadherin/catenin complex appears to occur in DSV via two different pathways: (1) E-cadherin alteration either through mutation or through methylation of the E-cadherin gene promoter and (2) β-catenin and/or γ-catenin alterations.
In a study of the genetic alterations in DSV, no BRAF (V600) mutations were found but all the cases showed RET proto-oncogene (RET)/PTC rearrangements.46 These rearrangements especially RET/PTC1 are more common in tumours seen in children and young adults; especially those associated with radiation exposure. The morphological features of DSV have been described in some of the thyroid tumours from children exposed to radiation after the Chernobyl accident.47 ,48 Therefore, it is not surprising that the presence of RET/PTC is a prominent genetic event in DSV-PTC and that these tumours are susceptible to radioactive iodine therapy. Another study supporting the above findings identified that the profile of genomic mutations detected in PTC-DSV is different from that in classic PTC.49 None of the genomic mutations of BRAF, 1p36, 3p12, 7p31 and 10q23 was detected in DSV, but a higher frequency of loss of heterozygosity of 3p24, 9p21, 17q21, 21q22 and 22q13 was noted. It has been suggested that these mutations may play an important role in the different morphological appearance and prognosis of DSV.
Solid variant
A solid growth pattern is noted focally in many papillary carcinomas. When the solid growth represents >50% of the tumour mass, a diagnosis of solid variant (SV) of papillary carcinoma may be made.4 The SV is most commonly seen in children and its incidence is 10% in sporadic PTC50 and >30% of patients with papillary carcinoma after the Chernobyl nuclear accident.51 It usually presents as a solid nodule with infiltration into the surrounding thyroid parenchyma. By light microscopy the tumour shows solid nests of tumour cells with nuclear features of papillary carcinoma, separated by delicate to broad collagenous bands (figure 4). Some tumours may show focal areas of follicular and papillary architecture. The nuclear features are those of papillary carcinoma, although the nuclei tend to be more rounded than oval. About 40% of these tumours show vascular invasion and ETE (figure 4).51

Solid variant: Solid nests of tumour cells showing nuclear features of papillary thyroid carcinoma (H&E stain, 40×).
Although some studies particularly from Japan have considered the solid papillary carcinoma as a poorly differentiated tumour with a guarded prognosis, in North America and Europe, this has not been found. It is important to recognise these lesions as papillary carcinomas and not overdiagnose them as more aggressive tumours such as poorly differentiated (insular) carcinoma.52 Papillary and non-papillary cancers of the thyroid can demonstrate areas of solid growth and/or insular growth patterns. We believe that these two should be morphologically separated; the areas of insular carcinoma usually show well-defined nests of monotonous tumour cells with round nuclei and scant cytoplasm. Loose connective tissue stroma and prominent vascularity separate the tumour nests from each other. Occasionally these areas may show foci of tumour necrosis.53 Albores-Saavedera et al described five cases of the macrofollicular variant of papillary carcinoma that exhibited a minor insular transformation. All these patients survived albeit with metastasis in 40%, these authors believed that the presence of the insular component did not alter the excellent prognosis associated with papillary carcinoma.54 Similar findings were reported by Ashfaq et al, who found that the aggressive clinical behaviour of tumours with focal insular change is more dependent on age of patient and tumour stage rather than insular growth pattern.55
SV does occur in adults and in our experience, over a third of these arise in patients with systemic autoimmune disease. The inter-relationship of these disorders is unknown. The prognosis of SV of papillary carcinoma is almost as good as classic papillary carcinoma, in children and in adults. They do not have the guarded prognosis of poorly differentiated carcinoma, even in the presence of necrosis.4 ,51
Molecular analysis of cases of SV-PTC arising in association with radiation exposure has shown RET/PTC rearrangement. The RET/PTC3 rearrangement has been more frequently associated with SV-PTC56 which is considered the most prevalent variant among the irradiated tumours. With the increase in latency period the prevalence of the SV and the RET/PTC rearrangements has declined suggesting a strong relationship between radiation exposure, SV and RET/PTC3.52 Interestingly, a triplet deletion of the BRAF gene leading to the replacement of a valine and a lysine by a glutamate (BRAF V600E+K601), was first reported only in the SV of PTC by Trovisco et al57; which has been confirmed by other investigators.58
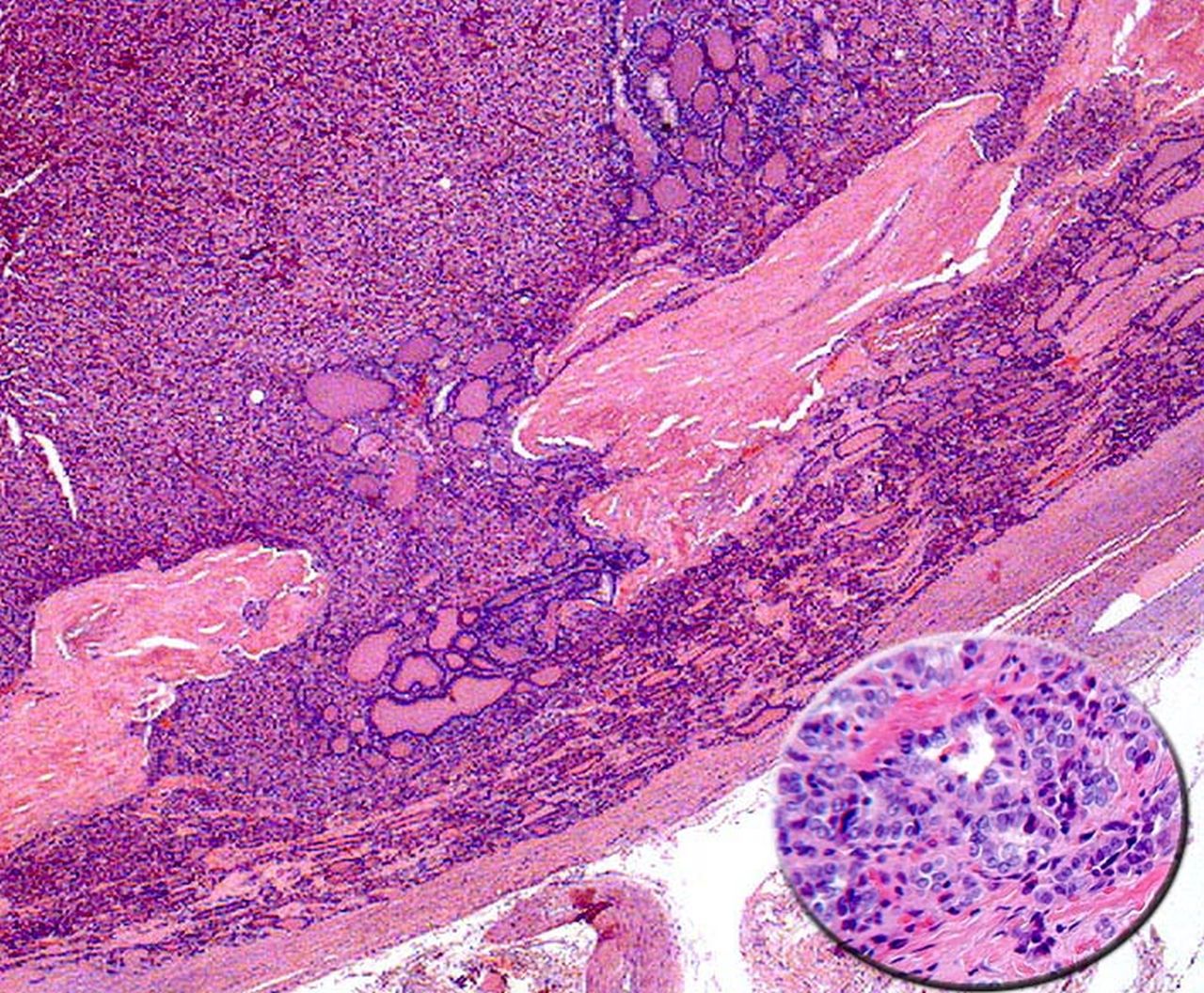
Hobnail variant
This rare variant of PTC was described by Asioli et al.59 These tumours commonly occur in women and are associated with significant mortality; 50% of the eight reported cases. The tumours in the reported series usually had more than 30% of the tumour with hobnail features. This peculiar feature is characterised by tumour cell nuclei located in the middle or in the apex of the cytoplasm that is, bulging of the nuclei at the tip of the cell imparting the so called ‘hobnail’ appearance to the cells (figure 5). This morphology is also encountered in serous papillary carcinoma of ovary, primary serous carcinoma of the peritoneum, breast micropapillary carcinoma, and bladder, kidney and lung adenocarcinomas with hobnail features.59 In a short communication Albores-Saavedra60 elegantly explained that these cases represent the rare oncocytic variant of papillary carcinoma based on the fact that similar cell morphology can be seen in oncocytic tumours as originally described by Rosai et al.61 In addition, these tumours demonstrate eosinophilic cytoplasm and electron microscopic studies show mitochondrial rich cytoplasm further supporting the oncocytic lineage. BRAF (V600E) mutations have been detected in >50% of these tumours confirming the classification of these tumours as variants of PTC.59

Hobnail variant: Tumour with papillary growth pattern; the inset shows some of the tumour cell nuclei are eccentrically placed causing bulging of the nuclei at the tip of the cell imparting the so-called ‘hobnail’ appearance to the cells (H&E Stain, 20× and 60×).
Widely invasive (diffuse) follicular variant of papillary thyroid carcinoma
The follicular variant of PTC (FVPTC) is characterised by possessing nuclear features typical of PTC (eg, nuclear clearing, grooves, and pseudoinclusions) and a follicular growth pattern. It is the second most common subtype of PTC, after classical PTC. Two distinct types of FVPTC include the common (or encapsulated) follicular variant with a less aggressive course and prognosis apparently similar to classical PTC, and the other widely invasive (diffuse) (or multinodular) follicular variant (diffuse FVPTC) with a more aggressive course.62 Widely invasive (diffuse) FVPTC is a distinct aggressive variant that occurs primarily in young women with increased multicentricity, extrathyroid extension, lymph node metastasis and venous invasion compared to uninodular, well-circumscribed, frequently encapsulated variant of FVPTC.63 Grossly, the tumour may resemble a multinodular goitre64 or fairly distinct whitish nodules dispersed throughout the thyroid parenchyma.62 Histologically, the tumour as defined by its name, has a multinodular or diffuse growth pattern involving one or both lobes of the thyroid gland with unencapsulated ill-defined or pushing borders. In contrast to the encapsulated variant that has predominance of medium-sized follicles, widely invasive (diffuse) FVPTC has predominance of microfollicles and/or trabeculae with frequent vascular invasion (figure 6). Recently, in a molecular analysis of four cases of widely invasive (diffuse) FVPTC, two cases (50%) had a BRAFV660E mutation whereas none had RAS mutations.65

Diffuse follicular variant of PTC: A solid and follicular growth pattern tumour demonstrating widespread invasion. The inset highlights the nuclear features of papillary thyroid carcinoma (H&E Stain, 20× and 60×).
Poorly differentiated thyroid carcinoma
Poorly differentiated thyroid carcinoma (PDTC) is a distinct diagnostic entity and its incidence is 2–3% of thyroid cancers in North America.66 The term was first introduced by Granner and Buckwalter in 196367 and then again defined in the 1980s by two different groups using two very different sets of criteria.17 ,53 The WHO finally acknowledged the entity of PDTC in 2004 as follicular cell derived neoplasms that show limited evidence of structural follicular cell architecture and occupy morphologically and behaviourally an intermediate position between differentiated and undifferentiated (anaplastic) carcinoma.3 Since, insular growth pattern to date is considered to be one of the hallmark features of PDTC it is prudent for the present discussion to describe ‘insular carcinoma’ of the thyroid. In 1984 Carcangiu et al described insular carcinoma as a tumour characterised by solid nests or islands (‘insulae’) of small and uniform tumour cells containing a variable number of microfollicles surrounded by thin-walled vessels (figure 7). The aggressive nature of this tumour was evident by presence of mitotic figures, foci of necrosis, and frequent lymphovascular invasion. Metastases to regional lymph nodes, lung and bones were common with high mortality rate as compared with conventional papillary and follicular carcinoma. These authors described the biological behaviour of this tumour as intermediate between well-differentiated and anaplastic thyroid carcinoma53 However, Sakamoto et al proposed that the term poorly differentiated carcinoma should be applied to the tumours that are solid or trabecular and have loss of follicular and/or papillary architecture.17 These variable definitions led many authors to even classify some of the aggressive variants of papillary carcinoma such as TCVs and CCVs as PDTC.68–71

Poorly differentiated carcinoma. Pleomorphic tumour cells demonstrating solid and invasive growth (H&E stain, 40×).
In 2006, a meeting was held in Turin to standardise the diagnostic criteria for PDTC. According to this consensus the poorly differentiated carcinoma is defined as a tumour with solid/trabecular/insular growth pattern, absence of classic nuclear features of papillary carcinoma, and presence of either convoluted nucleus or necrosis or mitotic activity (≥3 mitoses per 10 hpf). This consensus was in accordance with the WHO definition of PDTC.72 To further clarify, if solid, trabecular or insular growth is absent, the tumour is excluded from PDTC. If solid, trabecular or insular growth is present, but the tumour shows predominantly nuclear features of classical PTC, the tumour is excluded. If no features of classical PTC are identified, the tumour is assessed for the presence of either convoluted nucleus or necrosis or mitotic activity. Any or all of these features is then sufficient for a diagnosis of PDTC. If not, the lesion is deemed to be a well-differentiated follicular thyroid carcinoma. Encapsulation is not a diagnostic feature.73 However, Turin recommendations fell short of defining the amount of poorly differentiated areas within a thyroid carcinoma to render a diagnosis of PDTC. Some authors have shown that a minor insular component within a well-differentiated carcinoma does not affect the prognosis, while others have shown that a major insular component does alter the prognosis adversely. In a study by Dettmer et al even a minor component of poorly differentiated (PD) (10% to 50%) could adversely affect the prognosis.74 Many PDTC will show a relationship to a well differentiated carcinoma. This may be identified in the lesion at diagnosis of PDTC or may be found in previously resected tumours (ie, the patient has a history of a thyroid carcinoma and the poorly differentiated component is found in a recurrence or metastatic site. The well-differentiated tumour may be papillary carcinoma or a variant thereof (often follicular variant) or true follicular carcinoma.53 ,75 ,76
The diagnosis of PDTC cannot be made with certainty by fine needle aspiration cytology and is primarily made by histological examination. FNA smears are usually highly cellular with mostly isolated tumour cells and clusters of overlapping cells with microfollicles with colloid and can raise a possibility of PDTC.77 (figure 6C) Most notably, the PDTC have a striking resemblance to medullary carcinoma. Immunohistochemistry can be useful to avoid pitfalls as PDTC are thyroglobulin positive and calcitonin negative. Some studies have described a peculiar pattern of thyroglobulin staining as dot-like paranuclear deposits in most of the poorly differentiated carcinomas.66 ,78 In certain situations where diagnosis of poorly differentiated carcinoma is suspected on the basis of growth pattern, but characteristic convoluted nuclei, increased mitotic count or necrosis are lacking, use of p53 stain which is often positive and Ki67 which is increased (≥15%) may be helpful.66
Molecular profiling of PDTC is extremely heterogeneous. Different studies have found BRAF, KRAS and RET/PTC rearrangements. However, in a report describing molecular analysis of 65 cases of PDTC, RAS mutations in codon 61 were by far the most common genetic alteration in poorly differentiated carcinomas (23% of cases), with all mutations in NRAS except one in the HRAS gene. No KRAS, RET/PTC or PAX8/ PPAR γ genetic alteration was detected, and only a single BRAF mutation was found in a poorly differentiated carcinoma with a residual component of a TCV of papillary carcinoma.79 ,80
Anaplastic carcinoma (undifferentiated carcinoma)
Anaplastic carcinoma comprises only 1.7% of thyroid cancers in the USA81 with a decreasing trend with respect to the incidence of well differentiated thyroid cancer.82 It is an extremely malignant neoplasm and is usually fatal (mortality rate at 1 year 90%).83 ,84 Its incidence typically peaks at the 6–7th decade of life with median survival from time of diagnosis being 3 months.85 Despite the use of multimodality treatment combining surgery, external beam radiation and chemotherapy, long term survival is possible for selected patients only.86
From a clinical standpoint, a rapidly enlarging thyroid mass that is firm and fixed to surrounding structures in an elderly patient is quite suggestive for anaplastic thyroid carcinoma. The thyroid gland has often been enlarged for years, containing multiple nodules or a low-grade, well-differentiated carcinoma that has grown slowly. Recent rapid growth with tracheal compromise is often noted. Clinically and surgically these cancers usually infiltrate and replace the thyroid parenchyma and the juxtathyroidal tissues. Metastases to the regional lymph nodes and lungs are common.83 ,84 Because of its aggressive behaviour, all anaplastic thyroid tumours are classified as stage IV, regardless of tumour size, location or metastasis.87
Grossly, the neoplastic tissue is pale, firm or hard, and opaque. Foci of haemorrhage and necrosis are frequent. Foci of calcification are rare, and occasionally, there are regions of metaplastic cartilage and/or bone.83 ,88 Varied histological patterns are present: (1) rounded to irregular medium-sized to giant-sized cells with eosinophilic cytoplasm and large or giant nuclei (often bizarre) with prominent nucleoli; (figure 8A) (2) fusiform (spindle) cells in a fascicular or storiform pattern; and (3) medium-sized to large-sized cells with squamoid characteristics. Some of these cells may have clear cytoplasm. The neoplastic giant cells may have a single nucleus or may be multinucleated. These various cellular types can be mixed together, and transitional forms can be seen. Bizarre nuclei, often vesicular, are common; large nucleoli may be present. There are numerous mitotic figures, and some are atypical. ‘Osteoclast-type’ giant cells of histiocytic origin exist in a few tumours.89

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Anaplastic carcinoma. Round to spindle shaped tumour cells with marked nuclear pleomorphism (arrows) arranged in solid growth pattern (A—H&E stain, 40×). Fine needle aspiration demonstrating pleomorphic tumour cells (long arrow) in a background of inflammatory cells; mostly neutrophils (short arrow) (B—Papanicolaou stain, 60×).
Neoplastic cells may replace some portions of vessel walls, and small clusters of neoplastic cells may extend into individual thyroid follicles. Polymorphonuclear leukocytes can infiltrate the tumour and can be large in number near the necrotic regions83; some tumours have a prominent eosinophilic leukocytic infiltrate in areas that are not necrotic; tissue culture studies indicate that these types of anaplastic carcinomas can secrete an eosinophil chemotactic factor.90
A paucicellular variant has been reported and must be distinguished from Reidel's disease and fibromatosis of the neck. Clues to this distinction include foci of hyper-cellularity, necrosis and abnormal mitoses. The sparse neoplastic cells have atypical nuclei.91
In all types of anaplastic carcinomas, immunoreactivity to thyroglobulin is typically absent; when present, it is evident only in some larger ‘epithelioid’ cells. Often it is only noted at the invasive edges of the tumour which indicates false positivity due to diffusion from destroyed thyroid follicles. Immunoreactivity to keratin may be demonstrated and is the most common marker that suggests epithelial characteristics; however, different cytokeratins (by molecular weights) are expressed in some tumours and many examples show no cytokeratin immunoreactivity.92–94 Approximately 30% of anaplastic carcinomas express thyroid transcription factor-1; this marker if positive is extremely helpful in diagnosis of anaplastic carcinoma especially in small biopsies.95 Interpretation of immunostains must be done with caution because normal thyroid epithelium and remnants of well-differentiated carcinoma (or a benign nodule) may be trapped within the aggressive neoplasm.
Evidence of a previous nodular goitre or a follicular, Hurthle or papillary carcinoma may be often found if multiple sections of the neoplasm are taken. The tumours with substantial spindle-cell or giant-cell and spindle-cell components may be mistaken for soft-tissue neoplasms, but they are usually not recognisable as one of the well-characterised sarcomas.83 ,84 ,96 ,97
Most so-called ‘small-cell anaplastic carcinomas’ diagnosed in the past were malignant lymphomas. Small-cell carcinoma does exist but it is usually a medullary or poorly differentiated carcinoma and can be appropriately diagnosed with judicious use of immunostains.88 ,92 ,98
In middle-aged or elderly patients, a portion of an otherwise well-differentiated carcinoma may be anaplastic carcinoma, which has grave prognostic implications. If such a focus is only a few millimetres in diameter, it may have little effect on the patient's long-term survival, but in some patients, this is unfortunately not true.99–101 The same concept applies for a tiny anaplastic carcinoma discovered in a thyroid that was removed for multinodular goitre.
The cytology of anaplastic carcinoma shows marked cellularity or necrosis and haemorrhage, depending on the part of the mass sampled.102 ,103 Leukocytes can be numerous. Spindled and giant cells are present 104; with bizarre neoplastic cells with one or several nuclei (figure 8B). Abnormal mitotic figures may be seen. The smears may show cells from a follicular neoplasm or papillary carcinoma if one coexists with the anaplastic carcinoma.105 Therefore, this possibility illustrates the requirement that several aspirations should be performed when a fast-growing mass is present. Also, these neoplasms may be haemorrhagic or fibrotic; thus, the epithelial cells may be sparse or diluted by blood.
Molecular targets of anaplastic thyroid carcinoma that determine its highly aggressive nature remain unidentified. It has been demonstrated that a set of micro-RNA (miRNA) are significantly downregulated in anaplastic thyroid carcinoma with respect to normal thyroid tissue and to differentiated thyroid carcinomas especially miR-25 and miR-30d. These miRNA target the polycomb protein enhancer of zeste 2 (EZH2) that has oncogenic activity and is drastically upregulated in anaplastic thyroid carcinomas but not in the differentiated ones.106 An inverse correlation between the expression of these miRNA and EZH2 protein levels has been found thus postulating that the downregulation of miR-25 and miR-30d could contribute to the process of thyroid cancer progression, leading to the development of anaplastic carcinomas targeting EZH2 mRNA.107
Conclusions
The recognition of the aggressive follicular derived carcinomas by pathologists is important due to their clinical behaviour and prognosis. Thus, more robust universal definitions need to be applied in the diagnosis of these tumours to circumvent issues related to their misclassification and underdiagnosis. As reviewed above, these tumours are commonly associated with distant metastases and tumour recurrences and some such as poorly differentiated carcinoma and anaplastic carcinoma may not be amenable to standard treatment options that is, thyroidectomy and radioactive iodine ablation. In the past decade molecular studies have provided insight into the biology of rare aggressive follicular derived carcinoma which proves to be equally important in the diagnosis and treatment of these tumours.
Key messages
-
There are significant clinical consequences to the pathological diagnosis of TCV-PTC.
-
Any foci of tall cells should be mentioned in a pathology report regardless of the percentage of tall cell cytology found.
-
Molecular profiling of PDTC is extremely heterogeneous.
-
The recognition of the aggressive follicular derived carcinomas by pathologists is important due to their clinical behavior and prognosis.
References
Footnotes
-
Contributors All the authors have contributed equally in the preparation of this manuscript. VAL and ZB suggested the concept of the review and title. RT performed literature search and wrote the first draft of the review. ZB and VAL edited and generated the final draft of the manuscript. This was approved by all authors before submitting it to the Journal of Clinical Pathology.
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.