Article Text

Abstract
Aims Previous estimates of the prevalence of mismatch repair (MMR) deficiency and Lynch syndrome in small bowel cancer have varied widely. The aim of this study was to establish the prevalence of MMR deficiency and Lynch syndrome in a large group of small bowel adenocarcinomas.
Methods To this end, a total of 400 small bowel adenocarcinomas (332 resections, 68 biopsies) were collected through the Dutch nationwide registry of histopathology and cytopathology (Pathologisch-Anatomisch Landelijk Geautomatiseerd Archief (PALGA)). No preselection criteria, such as family history, were applied, thus avoiding (ascertainment) bias. MMR deficiency status was determined by immunohistochemical staining of MMR proteins, supplemented by MLH1 promoter hypermethylation analysis and next generation sequencing of the MMR genes.
Results MMR deficiency was observed in 22.3% of resected and 4.4% of biopsied small bowel carcinomas. Prevalence of Lynch syndrome was 6.2% in resections and 0.0% in biopsy samples. Patients with Lynch syndrome-associated small bowel cancer were significantly younger at the time of diagnosis than patients with MMR-proficient and sporadic MMR-deficient cancers (mean age of 54.6 years vs 66.6 years and 68.8 years, respectively, p<0.000).
Conclusions The prevalence of MMR deficiency and Lynch syndrome in resected small bowel adenocarcinomas is at least comparable to prevalence in colorectal cancers, a finding relevant both for treatment (immunotherapy) and family management. We recommend that all small bowel adenocarcinomas should be screened for MMR deficiency.
- intestine
- small
- gastrointestinal neoplasms
- genetics
- neoplastic syndromes
- hereditary
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Small bowel cancer is a rare form of cancer, with an incidence of less than 1.0/100 000,1 and little is known about the risk factors for development of this rare disease. However, monogenic cancer predisposition syndromes, such as familial adenomatous polyposis (FAP) and Lynch syndrome, are known to be responsible for a proportion of small bowel adenocarcinomas.2 While FAP, which is caused by a germline pathogenic variant in the APC gene, is characterised by the presence of polyposis coli, Lynch syndrome may be harder to recognise.3 4
Lynch syndrome is caused by germline pathogenic variants in one of four mismatch repair (MMR) genes (MLH1, MSH2 (EPCAM), MSH6 and PMS2) and predisposes carriers to the development of mainly colorectal and endometrial cancer.4 In addition, risk for several other malignancies is increased, including risk for small bowel adenocarcinomas, currently estimated to be between 0.4% and 12% for MLH1 and MSH2 variant carriers.5 Unlike FAP, there are no overt clinical characteristics that distinguish a small bowel malignancy in a patient with Lynch syndrome from a sporadic case, although a personal or family history of a Lynch syndrome-associated cancer may be suggestive. Surveillance of the duodenum is generally not recommended in Lynch syndrome due to lack of evidence supporting its effectiveness.6 Nonetheless, identification of a Lynch syndrome family via a small bowel cancer case may provide the patient and other family members with the opportunity for surveillance of the colon, which has proven value as a screening strategy.7 8
A hallmark of Lynch syndrome-related tumours is the presence of MMR deficiency, which results from biallelic inactivation of one of the MMR genes and can be demonstrated by immunohistochemical staining of tumour tissue for the MMR proteins and/or microsatellite instability (MSI) analysis.9 10 Lack of nuclear staining of neoplastic cells or presence of MSI is indicative of MMR deficiency. MMR deficiency in Lynch syndrome occurs due to a second somatic hit in neoplastic cells, in addition to a germline variant. MMR deficiency may also occur in sporadic cases due to somatic inactivation of both alleles.11 The presence of MMR deficiency might also be relevant to patient treatment, given that Programmed Death-Ligand 1 (PD-L1)-blockers produce a good response in MMR-deficient (colorectal) cancers regardless of sporadic or hereditary aetiology.11 12 Universal screening for MMR deficiency in small bowel cancers, as introduced for colorectal cancer and endometrial cancer in many countries,13 14 may therefore be warranted. The potential benefit of a comparable screening strategy can only be accurately assessed if the prevalence of MMR deficiency and Lynch syndrome in unselected small bowel cancer is first reliably estimated. Previous estimates of the prevalence of MMR deficiency were based on small cohorts and consequently showed wide variability (0%–35%).2 15 Few data are available on the prevalence of Lynch syndrome in these cohorts. In this study, a large, unbiased collection of small bowel cancers was used to reliably establish the prevalence of MMR deficiency and Lynch syndrome in this rare tumour group.
Methods
Cohort
The nationwide network and registry of histopathology and cytopathology in the Netherlands, also known as Pathologisch-Anatomisch Landelijk Geautomatiseerd Archief (PALGA), were consulted in 2017 in a nationwide search of tumour samples from patients with small bowel cancer.16 All excerpts labelled by the reporting pathologist as a neoplasm of the small bowel were extracted for the 5-year period, 2012–2016. The conclusions of the resulting pathology reports were then screened for:
All resected primary small bowel adenocarcinomas within the 5-year time frame. This resulted in the selection of 411 eligible tumour specimens.
The hundred most recent samples that included a biopsy of an adenocarcinoma with a (possible) primary origin in the small bowel. This second category of samples was added to ensure inclusion of unresectable cases (some duodenal adenocarcinomas present at an advanced stage and are not resectable due to the high morbidity of surgery).
Formalin-fixed paraffin-embedded (FFPE) material representative of these adenocarcinomas was then requested. Material from 332 resection specimens and 68 biopsy samples was obtained. Due to the anonymous nature of the samples and the rules and regulations of the PALGA network, obtaining consent was not possible or required.
Study procedures
The study flow is visualised in figure 1. On receipt, 4 µm sections were taken from the FFPE blocks and subjected to H&E staining and immunohistochemical staining of the MMR proteins. Additionally, depending on tumour size and histology, 10 µm sections or punches from the tumour were taken for later DNA isolation. Guided by a matching H&E slide, the 10 µm sections were microdissected to enrich for tumour. All samples were coded for complete anonymity according to Dutch guidelines. Anonymous basic personal data (age at diagnosis and gender) were available for each patient, in addition to historical pathology reports. No other clinical data were available.

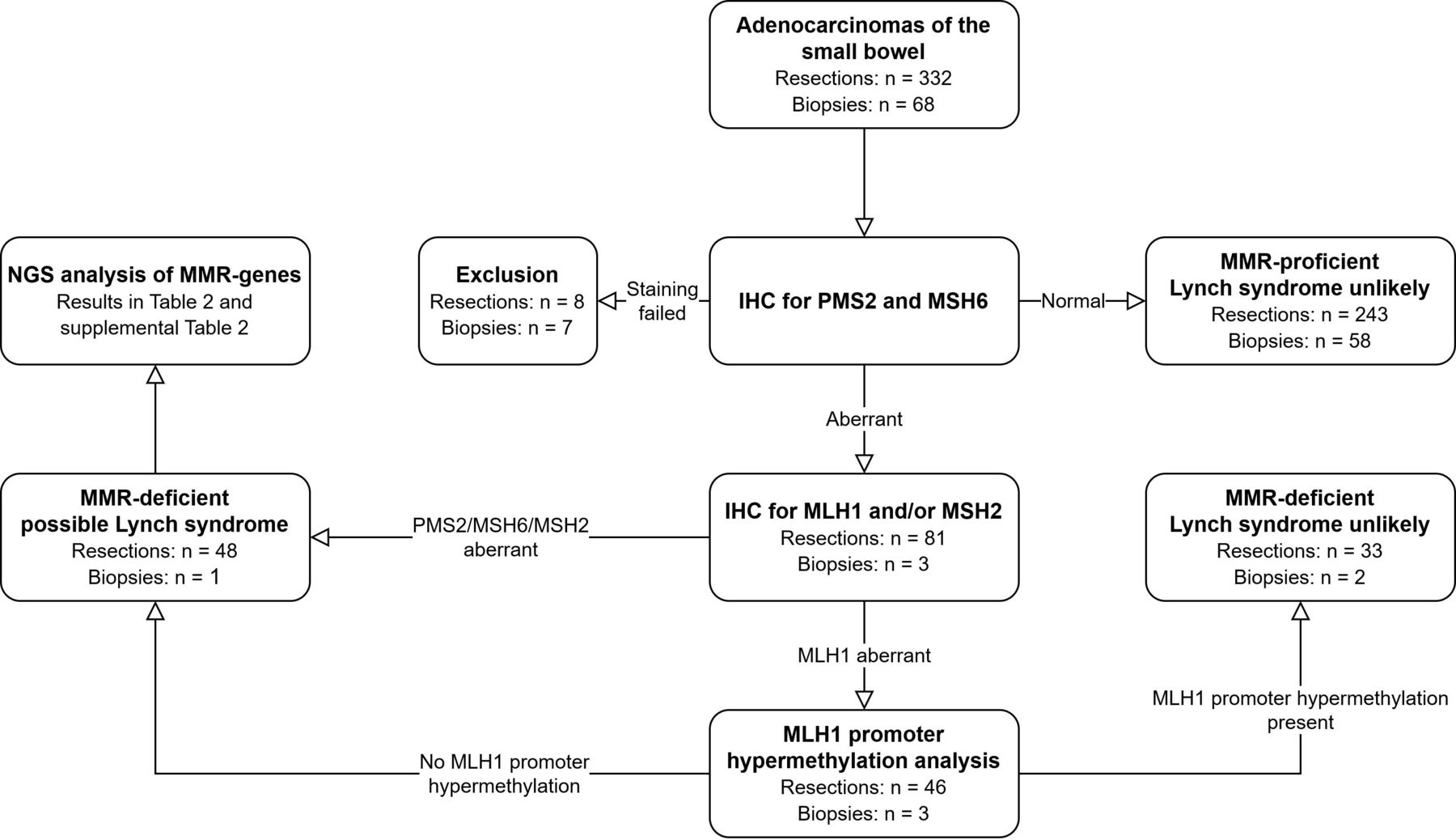{kind=link}
Study procedures. IHC, immunohistochemistry; MMR, mismatch repair; NGS, next generation sequencing.
All adenocarcinomas were initially immunohistochemically stained for PMS2 and MSH6 protein expression.17 Subsequent immunohistochemical staining for MLH1 and/or MSH2 was performed if the tumour was PMS2 deficient or MSH6 deficient. This approach is more cost-effective than using a four-antibody panel and has good sensitivity. The rationale for this approach is that functionally, MLH1 forms a heterodimer with PMS2, while MSH2 forms a heterodimer with MSH6, and mutations in MLH1 or MSH2 result in degradation of their heterodimer partners. Hence, use of PMS2 and MSH6 antibodies as a first screening step will generally identify loss of protein expression of MLH1 or MSH2.17 18 In cases with MLH1 deficiency, MLH1 promoter hypermethylation analysis was performed. In cases with loss of expression of MLH1 in the absence of MLH1 promoter hypermethylation or in cases with MSH2, MSH6 and solitary PMS2 expression loss, the MMR genes were further analysed using next generation sequencing (NGS). If NGS identified a variant with an allele frequency of >40%, DNA from matching non-neoplastic tissue (when available) was isolated to determine whether the variant was germline or somatic in origin.
Immunohistochemical staining
Details on the immunohistochemical staining procedures are found in the online supplemental methods. The immunohistochemically stained samples were examined by an experienced pathologist (HM or AFS) using light microscopy to evaluate MMR status. MMR proficiency was defined as the presence of nuclear staining within neoplastic cells as well as within adjacent non-neoplastic cells. MMR deficiency was defined as an absence of nuclear staining within neoplastic cells, together with positive expression in non-neoplastic cells. A third category, subclonal loss of protein expression, was defined for those adenocarcinomas harbouring a subpopulation of cancer cells with loss of expression together with cells retaining expression of an MMR protein.
Supplemental material
DNA isolation using the tissue preparation system
DNA was isolated using the Tissue Preparation System with VERSANT Tissue Preparation Reagents (Siemens Healthcare Diagnostics, Tarrytown, New York, USA), as previously described.19
MLH1 promoter hypermethylation analysis
Cases with loss of MLH1 expression were analysed for MLH1 promoter hypermethylation by methylation-specific PCR (MSP).20 21 Bisulphite conversion was carried out using the EZ DNA Methylation-Lightning Kit (D5031; Zymo Research) according to manufacturer’s instructions.
Targeted NGS
Adenocarcinomas with aberrant expression of at least one of the MMR proteins in the absence of MLH1 promoter hypermethylation underwent DNA variant analysis using an NGS panel. This panel consists of 20 colorectal cancer-associated and polyposis-associated genes and hotspot regions of the CTNNB1 gene (see online supplemental table 1 for all genes and panel coverage). For the purposes of this study, analysis of NGS results was restricted to MLH1, MSH2, MSH6 and PMS2. Sequencing was performed using the Ion Torrent platform according to the manufacturer’s recommendations. Details are found in the online supplemental methods.
Supplemental material
The unaligned sequence reads generated by the sequencer were mapped against a human reference genome (hg19) using the Burrows-Wheeler aligner. VarScan and ANNOVAR software were used for variant calling and annotation, respectively, and Integrative Genomics Viewer (IGV) software was used to visualise the read alignment and presence of variants. Additionally, the Leiden Open Variant Database, ClinVar and Alamut software were used whenever additional variant interpretation was needed.
Statistical analysis
Using IBM SPSS Statistics V.24, the χ2 test and one-way analysis of variance (ANOVA) test were performed as appropriate to compare patient and tumour characteristics of MMR-proficient cases with sporadic MMR-deficient cases and Lynch syndrome-associated cases. A p value<0.05 was considered to be statistically significant. Cases with subclonal loss of one of the MMR proteins were excluded from these analyses.
Results
Immunohistochemistry
The prevalence of MMR deficiency, as determined by immunohistochemical staining, was 22.3% in resected small bowel adenocarcinomas and 4.4% in biopsies (table 1). Additionally, seven (2.1%) resected samples showed subclonal loss of at least one MMR protein. Eight resected adenocarcinomas and seven adenocarcinoma biopsy samples had to be excluded from further analysis because no (representative) tumour tissue was present in the available FFPE blocks.
Prevalence of mismatch repair (MMR) deficiency and immunohistochemical staining patterns in resected and biopsied adenocarcinoma samples
Causes of mismatch repair deficiency
The most common cause of MMR deficiency was MLH1 promoter hypermethylation (40.5% of MMR-deficient resections and 66.7% of MMR-deficient biopsies, table 2). In more than a quarter of MMR-deficient resection samples, the MMR deficiency was related to Lynch syndrome (27%, table 2 and online supplemental table 2). The prevalence of Lynch syndrome within the total resection cohort was therefore at least 20/324 (6.2%). The true number might in fact be higher because in six cases an MMR gene variant with a high allele frequency (>40% of reads) was identified within the tumour, but matched normal tissue was not available to confirm or refute germline origin of the variant.
Supplemental material
Causes of mismatch repair (MMR) deficiency
Comparison of patient and tumour characteristics
A comparison of patient and tumour characteristics of MMR-proficient, (apparently) sporadic MMR-deficient and Lynch syndrome-associated cases included only the resected adenocarcinoma cases, as they represent the largest subcohort and have a documented primary tumour location within the small bowel. The six cases carrying a high allele frequency variant but without available-matched normal tissue were excluded due to uncertainty regarding their status as Lynch syndrome or sporadic MMR-deficient cases. Cases with an unexplained MMR deficiency and those with subclonal MMR deficiencies were also excluded from this analysis.
Mean age at cancer diagnosis was significantly lower in the patients with Lynch syndrome (table 3), and a previous history of a Lynch syndrome-associated cancer was significantly elevated in patients with Lynch syndrome. Interestingly, coeliac disease (diagnosed based on pathology reports of small bowel biopsies unconnected to the small bowel cancer diagnosis) was significantly more common in sporadic MMR-deficient cases. No other significant associations were identified (eg, location, gender, other cancer history,22 Crohn’s disease).
Cohort characteristics for Lynch syndrome versus mismatch repair (MMR) proficient versus MMR-deficient cases
Discussion
In a large group of resected primary small bowel adenocarcinomas, we found complete MMR deficiency in 22.3% and subclonal deficiency in 2.1% of cases, while biopsied small bowel adenocarcinomas showed a lower prevalence of MMR deficiency (4.4%). To the best of our knowledge, this is the first study to systematically screen a large, consecutive group of small bowel adenocarcinomas for the prevalence of MMR deficiency. Previous studies were either smaller and/or used selected cases with a higher a priori chance of being related to Lynch syndrome. Furthermore, many of these studies did not include molecular analysis to verify whether MMR deficiency was Lynch syndrome-related or sporadic.2 15 23
A recently published French study by Aparicio et al24 reported a Lynch syndrome prevalence of 6.9% in a large cohort of small bowel adenocarcinomas, in line with a prevalence of at least 6.2% in our cohort. MMR deficiency prevalence could not be compared because this French cohort was not systematically screened for MMR deficiency.
Of particular note, the prevalence of MMR deficiency in our study differed considerably between the resected and biopsied specimens. A higher prevalence of MMR deficiency in resected versus biopsied samples might be related to the association of MMR deficiency with a better prognosis in other cancers,25 so resections may represent patients with cancer with a relatively good prognosis, whereas biopsies may represent patients with a poor prognosis who are less likely to undergo resection. Interestingly, the prevalence of MMR deficiency identified in biopsied samples, 4.4%, is close to the 5.0% prevalence identified in a metastatic colorectal cancer cohort.26 However, as no further clinical data were available to verify that a biopsied sample was a confirmed primary small bowel cancer, our cohort may also have included cancers with a different primary location (where MMR deficiency prevalence is lower). Further validation of the prevalence of MMR deficiency in a cohort of small bowel cancers that were not resected is therefore required.
The relevance of subclonal loss of MMR protein expression is still poorly understood. While it seems unlikely that these patients have Lynch syndrome, the relevance of subclonal loss for prognosis and/or therapy will require further investigation.18 27
A significant overrepresentation of patients with coeliac disease was noted among cases with sporadic MMR deficiency. An association of coeliac disease with sporadic MMR deficiency (particularly with MLH1 promoter hypermethylation) has been described previously,28 29 and two out of three MMR-deficient cases from our cohort also showed MLH1 promoter hypermethylation. A limitation of our study was the lack of accompanying clinical data, which meant that we had no information on treatment/diet and could not verify whether the pathological signs of coeliac disease correlated with patient symptoms. These results should therefore be interpreted with caution, because there are other conditions that mimic the histological signs of coeliac disease.30
Another drawback of anonymous data is that it precludes verification of the number of Lynch syndrome cases, knowledge that might otherwise be used to establish how many patients are missed using current practices. Nevertheless, from pathology reports we could deduce that 13 out of 20 patients with Lynch were likely already identified, either because MSI and/or immunohistochemical testing was described (in the small bowel tumour or a previous tumour) or a previous diagnosis of Lynch syndrome was mentioned (online supplemental table 3).
Supplemental material
There is an ongoing discussion whether a two-antibody panel for immunohistochemical staining of the MMR proteins has sufficient sensitivity to detect MMR-deficient cases. Although a small number of MMR-deficient cases may be missed with a two-antibody panel, it is not expected that the results of a four-antibody approach would alter our conclusions.
A molecular cause of MMR deficiency could not always be identified (n=12). This is likely partly explained by the fact that we did not perform multiplex ligation-dependent probe amplification analysis to screen samples for deletions and/or insertions (germline or somatic) of the MMR genes or EPCAM (table 2 and online supplemental table 2). Nonetheless, NGS data were manually checked using the IGV for evidence suggesting a deletion, which led to the identification of deletions in three samples (online supplemental table 2, eg, study ID 33). Although this approach lowers the risk of missing copy number variants, not all deletions/insertions will be identified. As EPCAM was not sequenced, deletions of this gene will have been missed by definition. However, as only 1%–3% of all Lynch syndrome families carry an EPCAM deletion and deletions/insertions of the MMR genes explain a minority of Lynch syndrome families,4 31 Multiplex Ligation-dependent Probe Amplification (MLPA) analysis is unlikely to have altered our conclusions and recommendations. Another possible explanation for the failure of NGS results to resolve all MMR deficiency cases is that some cases lacked the informative single nucleotide polymorphisms required to determine whether loss of heterozygosity has occurred.
The analysis of PMS2 is complicated by the presence of pseudogenes. Nevertheless, researchers from our group have shown that it is possible to reliably detect variants in PMS2, even when using DNA isolated from FFPE material, as long as the correct amplicons are selected.32 Exceptions include variants in exon 12–15 due to gene conversion. The two germline variants identified in our cohort are found in exons 1–11.
In our cohort, the prevalence of MMR deficiency in resected cases (22.3%) was higher than the reported prevalence of MMR deficiency in colorectal cancer (15%).33 This finding has implications for daily clinical practice in relation to three important issues: prognosis, treatment and surveillance. In (early stage) colorectal cancer, MMR deficiency has been linked to a better prognosis,25 34 35 an association that may also hold true for MMR-deficient small bowel cancers. Indeed, the aforementioned study by Aparicio et al reported a trend towards better prognosis for Lynch-associated small bowel adenocarcinomas versus those related to Crohn’s disease.24 Furthermore, with the advent of immunoblockade therapy and its proven efficacy in MMR-deficient cancers,36 MMR status is relevant when formulating treatment strategies regardless of germline or sporadic status. Finally, due to the high prevalence of Lynch syndrome, small bowel cancer as an entity may facilitate the identification of new Lynch syndrome families and consequently allow surveillance measures to be offered.
In light of the high prevalence of MMR deficiency and Lynch syndrome, together with associated relevance and benefits, we recommend the implementation of universal screening of all primary small bowel adenocarcinomas for the presence of MMR deficiency. An age limit of 70 years is often used in the universal screening of colorectal cancers for mismatch repair deficiency. However, as the Lynch syndrome-associated cases included in our study showed a very broad age range (35–77 years, table 3) at diagnosis, we suggest that age limits on universal screening for small bowel cancer may be detrimental.
Take home messages
Prevalence of mismatch repair deficiency (22.3%) and Lynch syndrome (6.2%) in resected small bowel adenocarcinomas is high.
No clear clinical or histological predictors of mismatch repair deficiency were identified.
We recommend the implementation of universal screening for mismatch repair deficiency in all small bowel adenocarcinomas.
Acknowledgments
We thank Medactie.com for assistance with the editing of this manuscript. We thank our PALGA-group collaborators for providing patient samples: Dr E.J.M. Ahsmann, Klinische pathologie Groene Hart Ziekenhuis; Dr C. Jansen, Laboratorium Pathologie Oost-Nederland; R.S. van der Post, Radboud UMC Nijmegen; C. Wauters, CWZ Nijmegen; Dr C.Y. Yick, Amphia Ziekenhuis Breda.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Handling editor Runjan Chetty.
Twitter @ManonSuerink, @HristinaHr
Collaborators PALGA-group collaborators: Dr E J M Ahsmann, Groene Hart Ziekenhuis; Dr C Jansen, Laboratorium Pathologie Oost-Nederland; R S van der Post, Radboud UMC Nijmegen; C Wauters, CWZ Nijmegen; Dr C Y Yick, Amphia Ziekenhuis Breda.
Contributors MN, HM and TvW were the overall principal investigators in this study, they conceived the study and were responsible for the study design and supervision of the entire study. MN obtained financial support. MS and GK performed statistical analyses, interpreted the results and drafted the initial manuscript. MS, GK, DT, DvE, LS and HH performed laboratory experiments. MS, GK, LS and HH were responsible for sample preparation. HM and AFS assessed all pathology slides (H&E and immunohistochemical staining). AMJL assisted in the interpretation of the results, and reviewed and contributed to the writing of the manuscript. PALGA group provided patient samples. All authors approved the final report for publication.
Funding This work was supported by a grant from the Dutch Cancer Society (KWF UL 2012-5155).
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval A favourable ethical opinion was received from the Medical Ethical Review Board of Leiden University Medical Centre (reference number P16.313).
Provenance and peer review Not commissioned; internally peer reviewed.
Data availability statement Data are available upon reasonable request. Our data consist of deidentified participant data and are available upon reasonable request by contacting the corresponding author ([email protected]).